

Feynman e le proteine
Un nuovo approccio alla ricerca farmacologica
di Pietro Faccioli
 a.
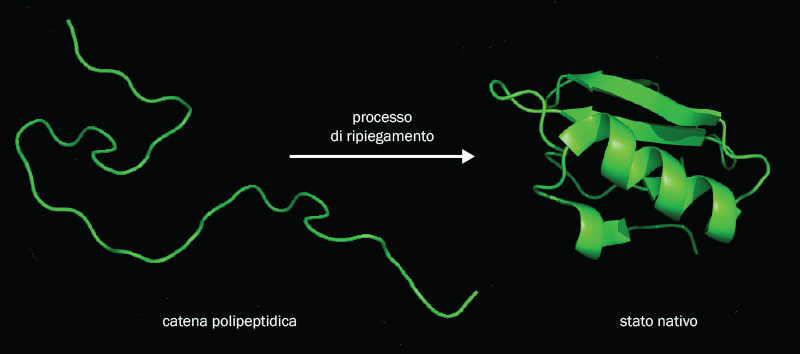
a. Rappresentazione schematica del processo di ripiegamento di una proteina.
I nuclei atomici e i loro costituenti sono sistemi di elevata densità, dominati dalla presenza di violente interazioni. Questa intrinseca complessità ha reso l’ambito della fisica teorica nucleare e subnucleare un contesto scientifico particolarmente adatto allo sviluppo di metodi di approssimazione e di tecniche numeriche avanzate. Nel corso di decenni, formalismi matematici, algoritmi, approssimazioni e persino macchine per il supercalcolo, originariamente concepiti per studiare questa fisica fondamentale, sono stati poi esportati con successo allo studio delle molecole e della materia condensata. Più recentemente, alcuni di questi metodi matematici sono stati impiegati per sviluppare nuovi algoritmi per la simulazione di un processo biologico fondamentale: il meccanismo di ripiegamento delle proteine. Questa innovazione ha ispirato un approccio completamente nuovo alla ricerca farmacologica, che sta portando alla scoperta di potenziali nuovi farmaci per malattie incurabili.
Le proteine sono catene polipeptidiche composte da 20 diversi tipi di aminoacidi. In generale, se la sequenza di aminoacidi è scelta in modo casuale, queste catene non assumono una forma precisa, ma cambiano struttura di continuo. Al contrario, le specifiche sequenze di aminoacidi che si trovano nelle proteine selezionate dall’evoluzione generano un comportamento completamente diverso: quasi tutte le proteine, infatti, assumono un’unica conformazione, in cui svolgono la loro azione biologica. Queste speciali catene sono in grado di ripiegarsi su se stesse spontaneamente, visitando stati via via più favorevoli energeticamente, fino a raggiungere la conformazione di minima energia, chiamata “stato nativo” (vd. fig. a).
Comprendere e caratterizzare con livello di dettaglio di singolo atomo il meccanismo di ripiegamento delle proteine avrebbe ovviamente enormi ricadute scientifiche e tecnologiche. In principio, simulazioni al computer permettono lo studio con risoluzione atomica di questo processo. In pratica, tuttavia, questi calcoli sono proibitivamente impegnativi.
La ragione di base è che i processi di ripiegamento sono eventi che avvengono molto raramente. Infatti, la catena passa la maggior parte del suo tempo alla ricerca casuale della “strada” che conduce allo stato nativo. Molto raramente, dopo innumerevoli tentativi sbagliati, la proteina “imbocca la strada giusta” e quindi raggiunge rapidamente lo stato nativo. Gli algoritmi tradizionali per la soluzione delle equazioni di Newton (chiamati di “dinamica molecolare”) richiedono di simulare tutti i tentativi falliti. Invariabilmente, il tempo computazionale a disposizione si esaurisce prima di riuscire a osservare anche solo un evento di ripiegamento.
 b.
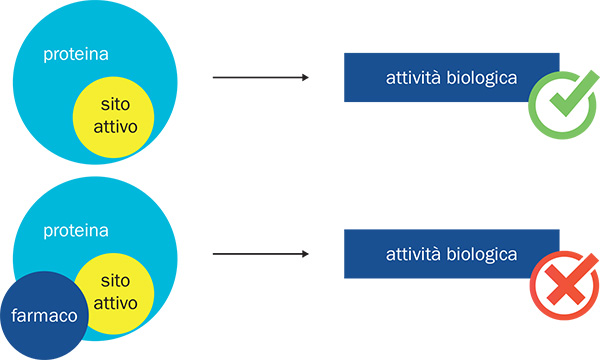
b. L’approccio convenzionale nella ricerca farmacologica razionale consiste nell’identificare piccole molecole in grado di legarsi alla superficie delle proteine bersaglio nella loro forma nativa, inibendone in questo modo l’attività biologica.
L’approccio razionale più comune per la scoperta di nuovi candidati a farmaci si basa sull’identificazione di piccole molecole, in grado di legarsi a specifiche proteine “bersaglio”, impedendo loro di contribuire a processi patologici (vd. fig. b). Purtroppo, alcune proteine sfuggono a questo tipo di “attacco”. Ad esempio, ciò può accadere quando la struttura nativa della proteina non presenta una regione concava in cui una piccola molecola può legarsi per poi interferire con la funzione biologica. In generale, le proteine che non si riescono a fermare con approcci farmacologici tradizionali vengono denominate “undruggable”.
Conoscere percorsi di ripiegamento delle proteine bersaglio ci ha consentito di concepire un approccio per rimuoverle dalla cellula, denominato PPI-FIT: Pharmacological Protein Inactivation by Folding Intermediate Targeting. L’idea di base è cercare piccole molecole che possano legarsi alla proteina bersaglio e immobilizzarla, prima che questa raggiunga lo stato nativo. Le cellule sono dotate di un meccanismo di controllo di qualità che consente loro di riconoscere ed eliminare proteine parzialmente o erroneamente ripiegate. In questo modo, le proteine bersaglio sono distrutte dalla cellula stessa.
 b.
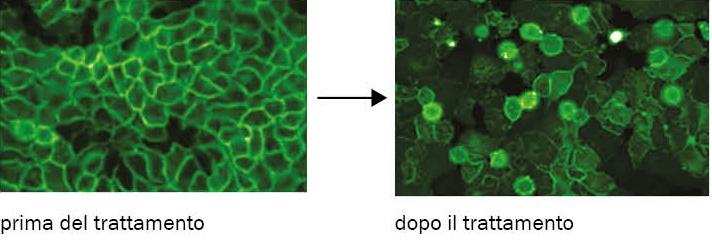
b. A sinistra: la molecola scoperta tramite PPI-FIT (SM875) riduce i livelli di concentrazione cellulare PrPc. A destra: dopo l’esposizione a SM875, le catene di PrPc non raggiungono più la membrana cellulare e si accumulano nei centri di degradazione della cellula.
Biografia
Pietro Faccioli ha conseguito il dottorato in fisica presso l’Università di New York a Stony Brook (USA) nel 2002, ha poi lavorato come ricercatore post-doc presso l’European Centre for Theoretical Nuclear Physics and Related Areas (Trento). È stato visiting scientist presso il MIT (USA) e l’Institute for Theoretical Physics del CEA (Parigi). Già professore in fisica teorica presso l’Università di Trento, è oggi professore di fisica applicata presso l’Università di Milano-Bicocca.
